PathoPic – image database / PathoPic ID 10588 - Hämophagozytose im Randsinus eines Lymphknotens
de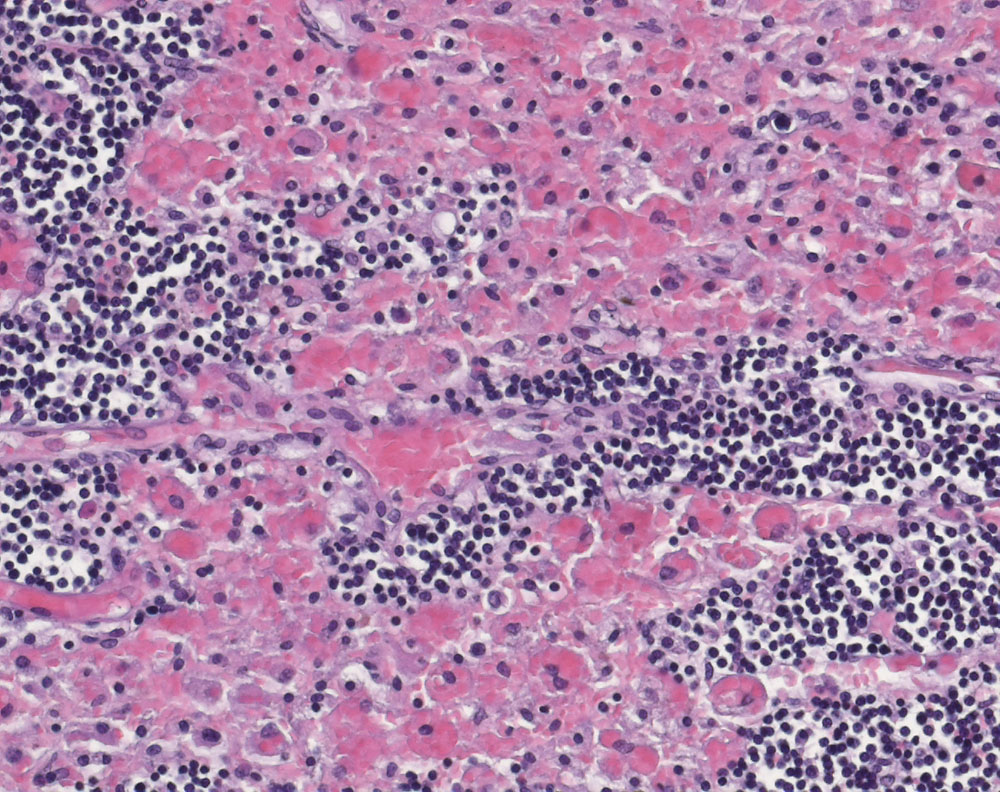
Diagnose
Hämophagozytose im Randsinus eines Lymphknotens
Diagnose Gruppe
Systemerkrankung/Immunpathologie
Topographie
Lymphknoten, mediastinal
Topographie Gruppe
Lymphatische Gewebe, KM, Milz
Beschreibung
Zahlreiche Sinushistiozyten enthalten phagozytierte Erythrozyten.
Klinik
SIRS bei Pneumonie links basal. St.n. hämorrhagischem Schock. Bizytopenie bei bekannter chronischer lymphatischer Leukämie. Leichte cholestatische Hepatitis.
Kommentar
Die histologischen Befunde passen zu einem hämophagozytischen Syndrom. Aufgrund der fehlenden Ferritin- und Interleukin-2-Rezeptorwerte ist dieses Syndrom anhand der histologischen Befunde allein nicht sicher diagnostizierbar. Die Aetiologie der Hämophagozytose ist sowohl klinisch, serologisch als auch durch immunhistochemischen Untersuchungen nicht CMV-, EBV- oder HIV-assoziiert. Am ehesten hat die Pneumonie in Zusammenhang mit der chronischen lymphatischen Leukämie die Hämophagozytose getriggert.
Bei den häufig verwendeten Richtlinien zur Diagnose des Hämophagozytose-Syndroms müssen minimal 5 von 8 der folgenden Kriterien erfüllt sein: 1) Fieber, 2) Splenomegalie, 3) Periphere Zytopenie in minimal 2 Zelllinien, 4) Hypertriglyzeridämie (Triglyzeride nüchtern 03,0 mmol/l) und/oder Hypofibrinogenämie (Fibrinogen 01,5 g/l), 5) Zeichen der Hämophagozytose im Knochenmark, in der Milz oder in Lymphknoten, 6) verminderte oder fehlende NK-Zell-Aktivität, 7) Ferritin 0500 mg/l und 8) soluble CD25 02400 U/ml. Zur Diagnose einer familiären Form reicht der molekulargenetische Nachweis einer pathognomonischen Mutation (z.B. Mutationen im Perforin-, Munc-, Syntaxin- oder SLAM-associated protein Gen), ohne dass die klinischen Diagnosekriterien erfüllt sein müssen.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Bei den häufig verwendeten Richtlinien zur Diagnose des Hämophagozytose-Syndroms müssen minimal 5 von 8 der folgenden Kriterien erfüllt sein: 1) Fieber, 2) Splenomegalie, 3) Periphere Zytopenie in minimal 2 Zelllinien, 4) Hypertriglyzeridämie (Triglyzeride nüchtern 03,0 mmol/l) und/oder Hypofibrinogenämie (Fibrinogen 01,5 g/l), 5) Zeichen der Hämophagozytose im Knochenmark, in der Milz oder in Lymphknoten, 6) verminderte oder fehlende NK-Zell-Aktivität, 7) Ferritin 0500 mg/l und 8) soluble CD25 02400 U/ml. Zur Diagnose einer familiären Form reicht der molekulargenetische Nachweis einer pathognomonischen Mutation (z.B. Mutationen im Perforin-, Munc-, Syntaxin- oder SLAM-associated protein Gen), ohne dass die klinischen Diagnosekriterien erfüllt sein müssen.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Bilder Typ
Histologie
Vergrösserung
200
Alter
86
Geschlecht
unbekannt
Datum
Ersteintrag: 20.05.2010
Update: 22.02.2011